

 Processing
Processing
Solid Tumor Research
Solid tumor oncology research has greatly benefited from next generation sequencing, but challenges remain, such as those with low-input samples, amplification of novel fusions, cumbersome workflows, and resource-heavy bioinformatic pipeline needs. Learn about these challenges and how Archer™ NGS assay solutions can help.
Archer NGS assay solutions
Overview
Solid Tumor Profiling Challenges with NGS
Solid tumor oncology research has greatly benefited from next generation sequencing methods to find relevant cancer-driving biomarkers. NGS’s ability to interrogate hundreds of genes for variants at very low levels has been a boon to molecular labs. The use of NGS for more complicated genomic alterations or assessments, such as structural variants, MSI, and TMB, has fueled comprehensive genomic profiling of solid tumor samples, ultimately providing labs with more information to understand these cancers.
However, traditional NGS methods may not provide all relevant information and can have challenging workflows and analysis. For example, traditional opposing primer assays limit identification of fusions to known partners only, missing potentially relevant novel fusions (Figure 1). Samples with low tumor cellularity, degraded nucleic acids (e.g. FFPE), or low-input (e.g. cfDNA) can all make detection of genomic alterations challenging.
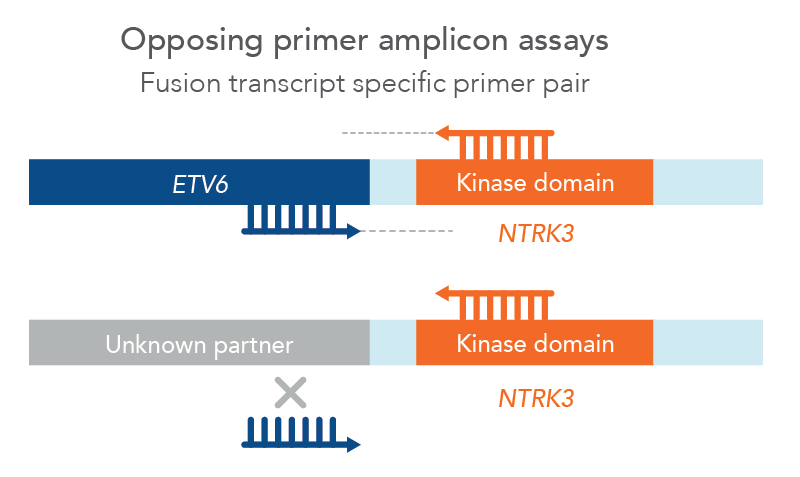
Figure 1. Opposing primer amplicon assays lack detection of unknown fusions. In this example, a primer pair for ETV6::NTRK3 can only amplify that specific fusion since the ETV6 primer cannot bind to an unknown partner, therefore missing a novel NTKR3 fusion.
Additionally, NGS library preparation workflows can be cumbersome, allowing many points for manual errors. Bioinformatic pipelines can be expensive and time-consuming to maintain. A pipeline may also lack the QC and statistical tools necessary to reduce errors and guide calling, which can leave it to lab staff to make difficult calls.
Archer NGS assay solutions for solid tumor research resolves these concerns with patented AMP™ chemistry, streamlined parallel workflows, and user-friendly analysis. Additionally, assays are designed to fit your lab’s unique needs from comprehensive profiling and high-throughput workflows, to smaller footprint assays and low-throughput manual workflows.
Detect more with AMP chemistry
Anchored Multiplex PCR (AMP) is a target enrichment method that uses molecular barcoded adapters and single, nested, gene-specific primers for amplification, permitting open-ended capture of DNA, RNA, and cfDNA fragments. This approach enables the detection of novel fusions (Figure 2), allows for flexible primer design, and protects against dropout of primers. MBC adapter ligation prior to PCR combined with Archer Analysis software power the correction of systematic errors and quantitative analysis of the original unamplified template.
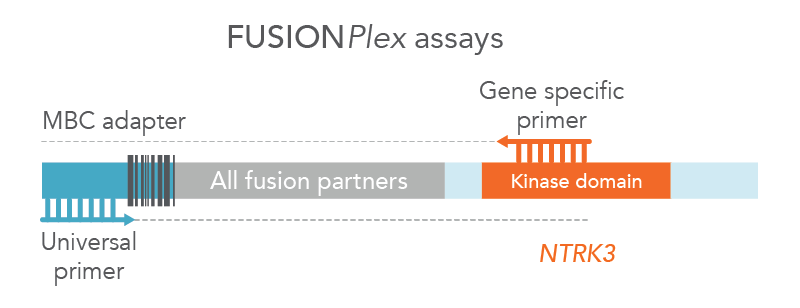
Figure 2. FUSIONPlex™ assays detect both known and novel fusions. FUSIONPlex assays include MBC adapters with a universal priming site. Since it is paired with the relevant gene-specific primer (in this case, NTRK3), both known and novel fusions are amplified.
High performance even with low input samples
Samples with low tumor cellularity, degraded nucleic acids (e.g. FFPE), or low input (cfDNA) that are typically challenging for traditional amplicon NGS variant detection are reliably amplified with AMP chemistry. In fact, 96% of expected fusions were identified with the FUSIONPlex Core Solid Tumor panel using 50 ng of 10% fusion positive material. (Figure 3)
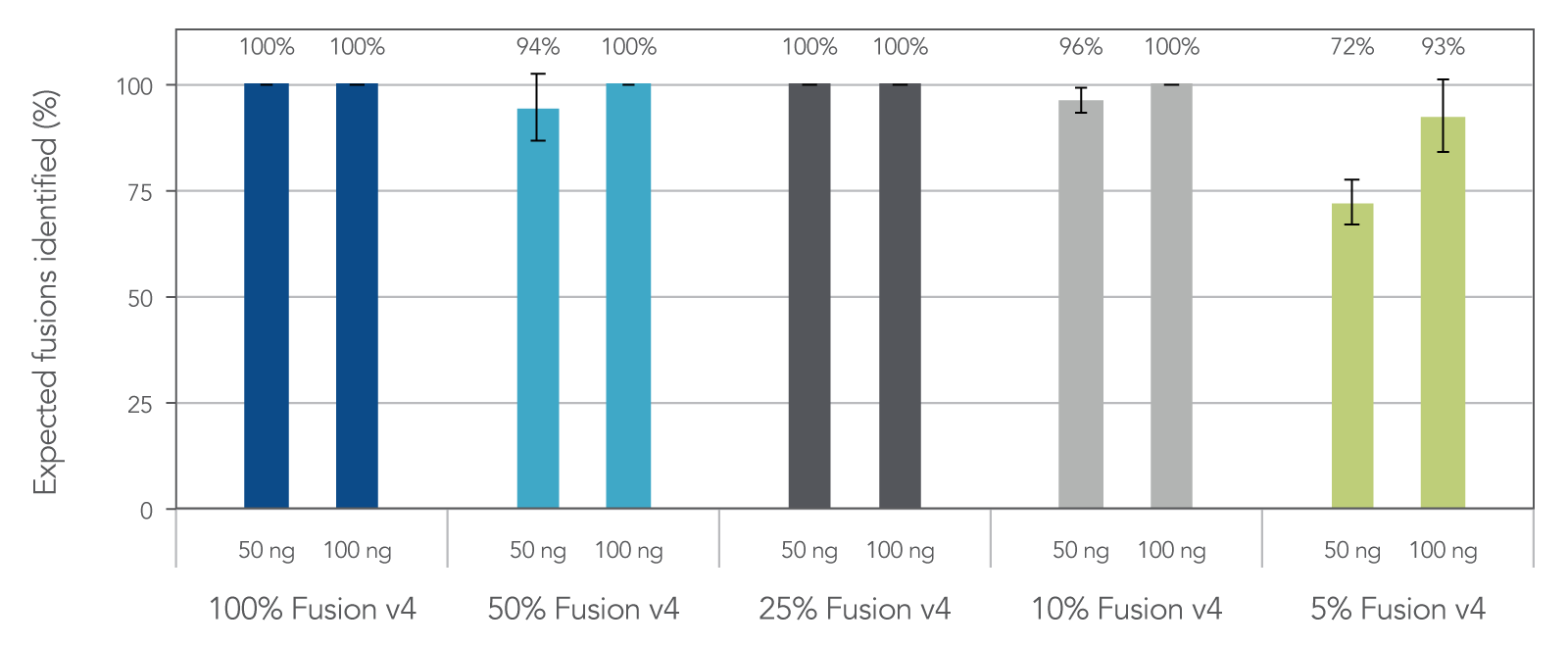
Figure 3. Sensitive fusion detection down to 5% fusion-positive reference material. Reference materials used: (1) Seraseq® FFPE Fusion RNA Reference Material v4 (0710–0496), (Background) Seraseq® FFPE WT (DNA/RNA) Reference Material (0710–0137), n = 3 per input mass, per dilution.
Genomic alterations, including fusions and CNVs, are consistently amplified from FFPE tissue with Archer solid tumor research assays. Figure 4 demonstrates one example of reliable variant calls by the VARIANTPlex™ Core Solid Tumor panel, with identification 100% of expected variants in FFPE reference material.
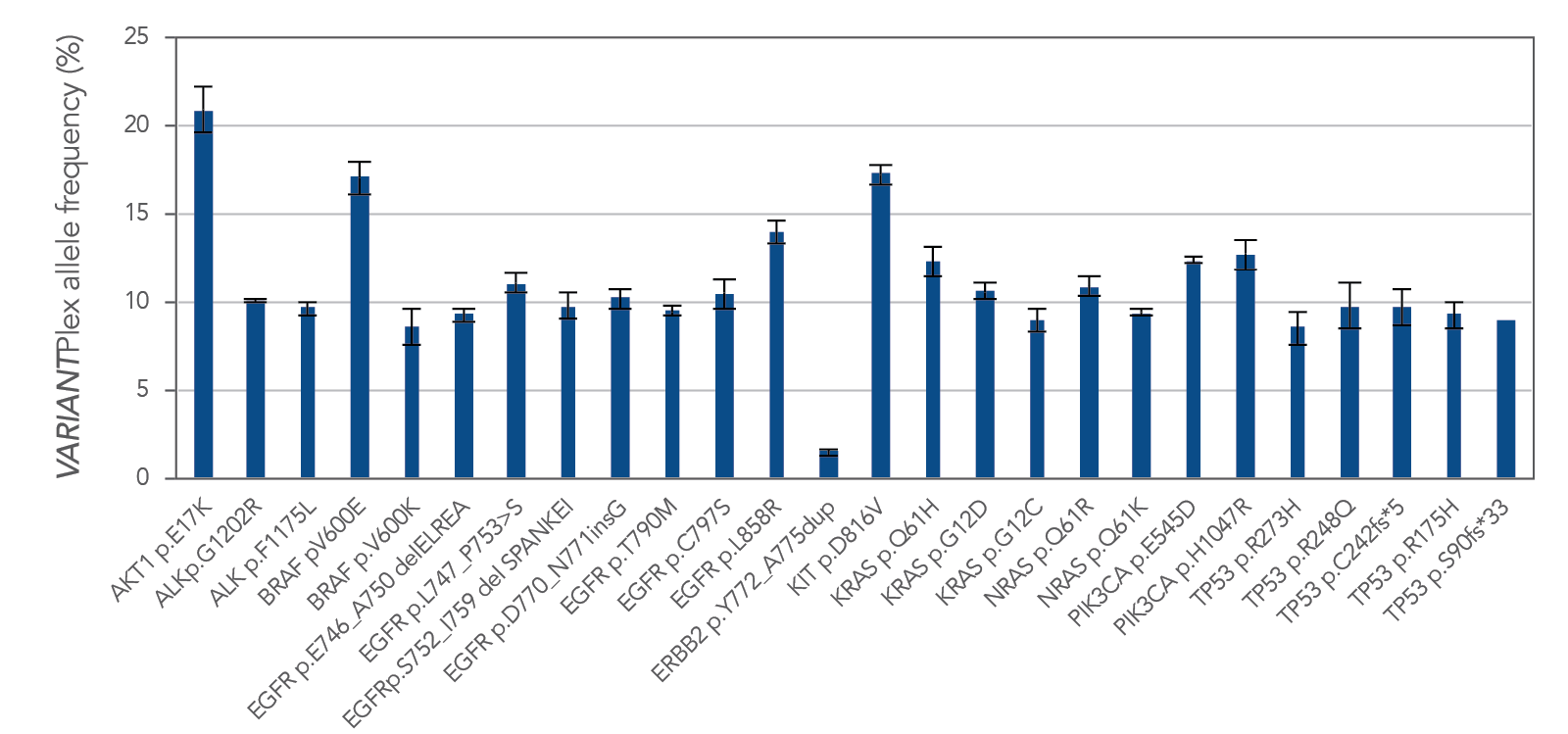
Figure 4. Variant calls by VARIANTPlex™ Core Solid Tumor, with 100% identification of expected variants. Reference material was Seraseq® Compromised FFPE Tumor DNA (0710–1492), n = 3, 50 ng input mass per library.
For solid tumor variant detection in plasma samples, AMP chemistry provides preferential enrichment of cell-free fraction compared to amplicon-based NGS. Additionally, Archer’s unique outlier identification algorithm leverages position-specific noise data to enable confident reporting at low allele frequencies.
Solid tumor panels and reagent formats fit for your lab’s needs
| Genes Targeted | Genomic alterations detected | Recommended reads | ||
|---|---|---|---|---|
| DNA Input | VARIANTPlex | |||
| Complete Solid Tumor | 430 | SNVs, indels, CNVs, ITDs, MSI, TMB | 45 M | |
| Pan Solid Tumor | 185 | SNVs, indels, CNVs, ITDs, MSI, TMB | 25 M | |
| Expanded Solid Tumor | 76 | SNVs, indels, CNVs, ITDs, MSI | 6 M | |
| Core Solid Tumor | 60 | SNVs, indels, CNVs, ITDs, MSI | 4.5 M | |
| Solid Tumor Focus v2 | 20 | SNVs, indels, CNVs, ITDs, MSI | 1.5 M | |
| RNA Input | FUSIONPlex | |||
| Pan Solid Tumor v2 | 137 | Fusions, SNVs, indels, expression | 3.5 M | |
| Core Solid Tumor | 57 | Fusions, SNVs, indels, expression | 3 M | |
| Sarcoma v2 | 63 | Fusions, SNVs, indels, expression | 2 M | |
| Lung v2 | 17 | Fusions, SNVs, indels, expression | 1 M | |
| ctDNA Input | LIQUIDPlex | |||
| Universal Solid Tumor | 29 | SNVs, indels | 5 M | |
VARIANTPlex Pan Solid Tumor and Complete Solid Tumor panels offer TMB in small and medium NGS panels with simultaneous detection of other relevant alterations, while demonstrating high concordance with WES-based TMB, as shown in a scientific poster presented at AMP 2023.
Additionally, the IMMUNOVerse TCR panel can provide tumor-infiltrating lymphocyte information relevant for solid tumor characterization.
Pair DNA- and RNA- based assays to efficiently generate a complete biomarker profile
| Paired Panels | Genes | Reads | Samples* per NextSeq 500/550 high output kits v2.5 |
|---|---|---|---|
| FUSIONPlex Lung v2 | 17 | 1M | 160 |
| VARIANTPlex Solid Tumor Focus v2 | 20 | 1.5M | |
| FUSIONPlex Core Solid Tumor | 57 | 3M | 53 |
| VARIANTPlex Core Solid Tumor | 60 | 4.5M | |
| FUSIONPlex Pan Solid Tumor v2 | 137 | 3.5M | 14 |
| VARIANTPlex Pan Solid Tumor | 185 | 25M | |
| FUSIONPlex Pan Solid Tumor v2 | 137 | 3.5M | 8 |
| VARIANTPlex Complete Solid Tumor | 430 | 45M |
Data assumes Archer libraries are prepared using the liquid adapters.
*Sample = 1 VARIANTPlex library + 1 FUSIONPlex library
VARIANTPlex and FUSIONPlex panels can be paired for cost-effective solutions to provide SNVs, indels, ITDs, fusions, and expression alterations from a single sample.
Choice of assay reagent formats are available to suit your lab’s sample volumes.
- For high-throughput labs, liquid reagents are ideal for automated liquid handling workflows. Liquid adapters are available in a 96-well plate format and include an increased number of dual indexes (over 36,000 unique combinations of sequencing indices) to support high-throughput sequencing.
- For manual workflow labs, single-use, reaction-size lyophilized reagents allow your lab to avoid potential contamination during manual pipetting and provide extended shelf life.
Both reagent formats have demonstrated excellent performance, with identification of 100% of expected variants in reference materials*. Regardless of reagent format, all reagents are color-coded to match protocol steps, making benchwork easy (Figure 5).
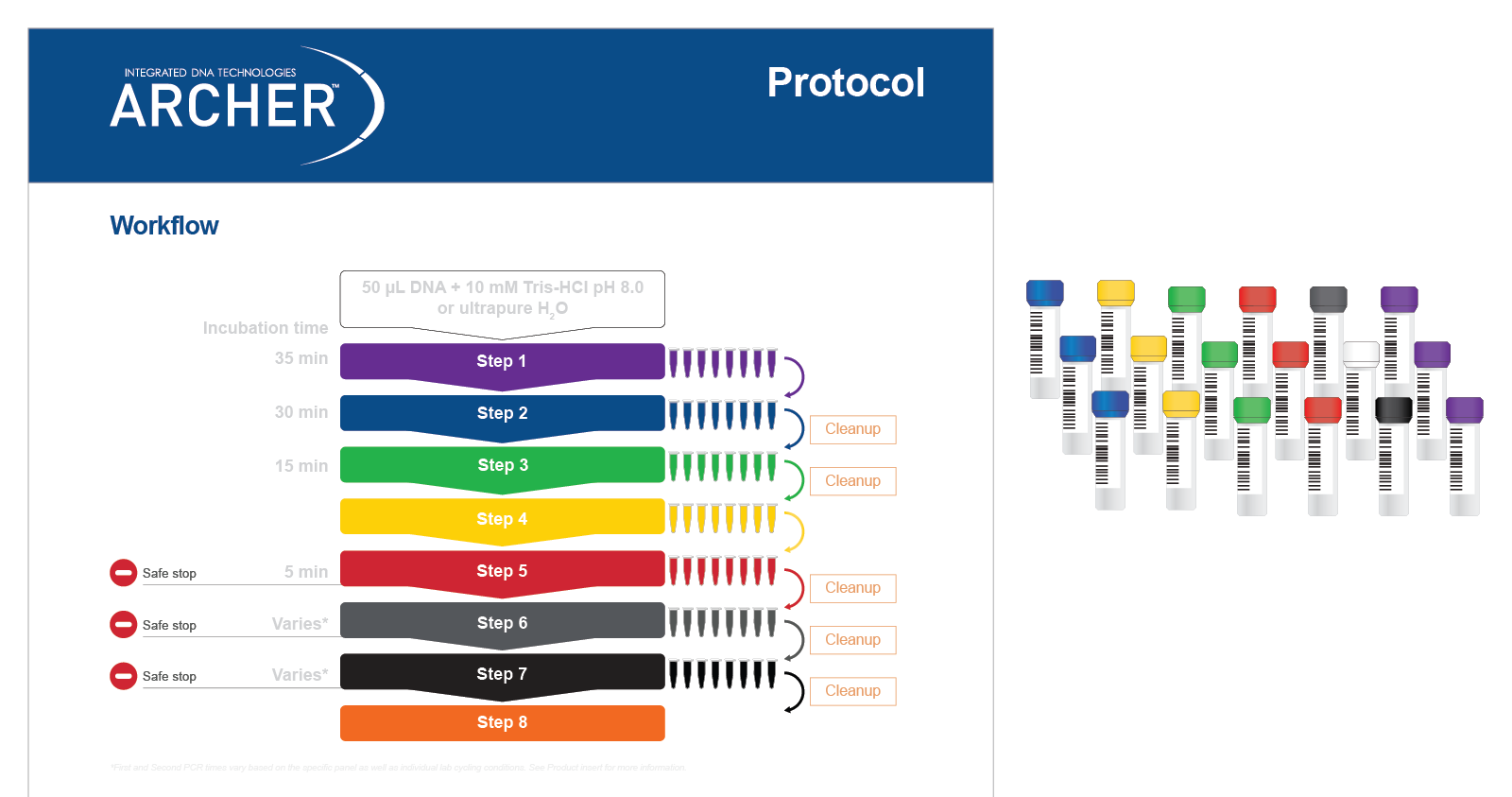
Figure 5. Reagents are color-matched to protocol steps for easy benchwork. Each step of the protocol has a different color, which matches the packaging of the reagents (in this case liquid reagents) for that step.
Customize your panel content
Relevant oncology biomarkers are emerging rapidly, necessitating labs to modify their assays to meet the pace of discovery. With the foundation of proprietary AMP chemistry and Archer Analysis you can confidently add targets to existing Archer assays or start from scratch without compromising assay performance. To get started, visit Assay Marketplace, the Archer assay design tool.
*Data available upon request
Solid Tumor Research Assays
Comprehensive Genomic Profiling
Targeted Pan-Cancer Panels
Tumor-Specific Panels
Other solid tumor panels
Ready to start?
Learn how our solid tumor research assays can identify key genomic alterations for your research.
Request a consultationResources
Posters


Videos
RUO23-2454_001